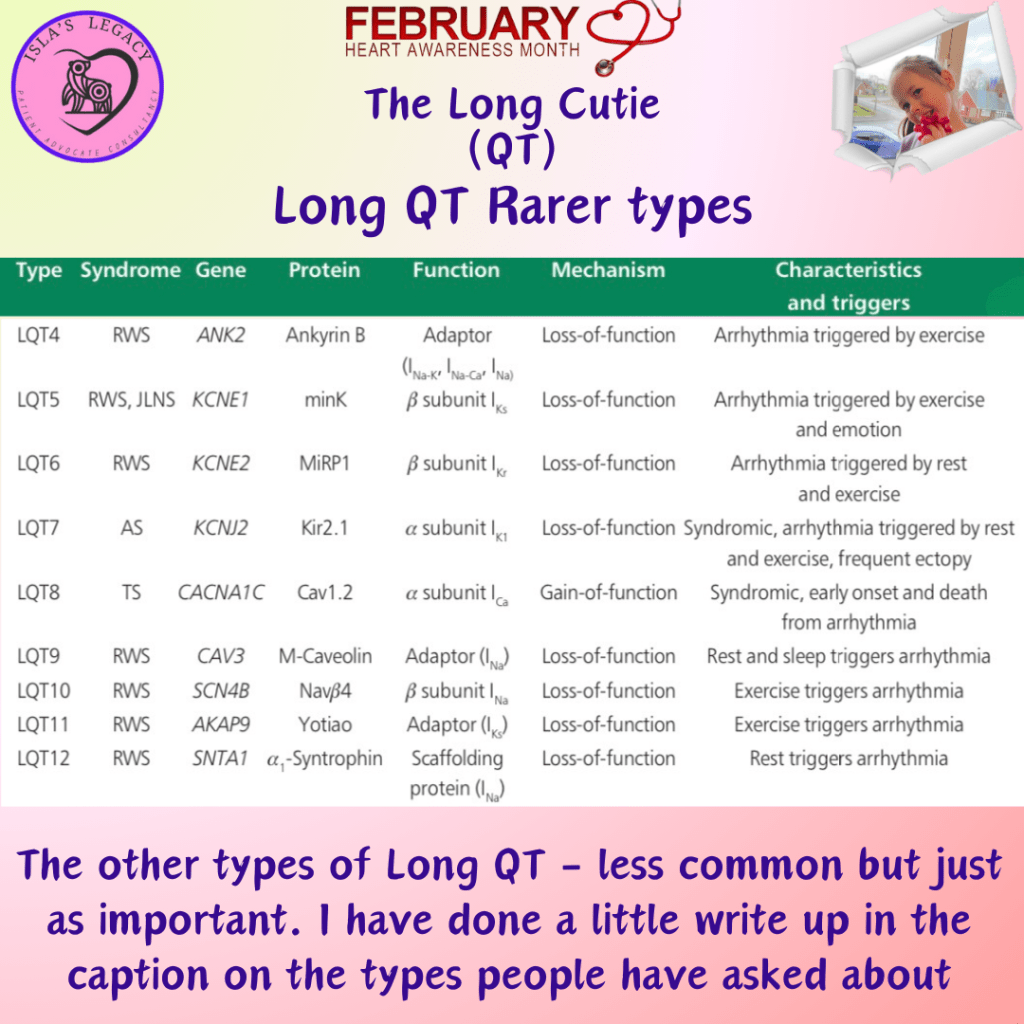
Some information on some of the rarer types of Long QT. Interestingly one of these types may actually be no longer considered as being a Long QT mutation… more on that tomorrow.
If anyone would like more on any other types or have any Long QT questions please reach out ☺️
Type 4 is a rare variety of LQTS, accounting for nearly 1% of cases. The affected gene is ANKB which encodes ankyrin-β, a structural protein. Mutations causing a loss of ankyrin-β function lead to increases in intracellular calcium concentration giving rise to early and delayed after-depolarizations. Arrhythmias are due to spontaneous depolarizations, usually in response to catecholaminergic stimulation (Included among catecholamines are epinephrine (adrenaline), norepinephrine (noradrenaline), and dopamine).
Research and related insights into Long QT Syndrome Type 5 (LQT5) is relatively new in the field, and prior to late 2019/early 2020 had been limited. LQT5 accounts for only 1-2% of all long QT syndrome cases and is caused by loss-of-function of a potassium channel coded for by KCNE1. While it isn’t known if beta blockers provide protection, they are recommended for KCNE1 positive mutations alongside avoiding QT prolonging drugs.
Type 8 arises from mutations in the CACNA1 gene which codes for L-type calcium channel Cav1.2. Because the CACNA1C gene was the eighth gene proved to cause QTc prolongation, it was historically called LQT8, but today a clear distinction exists between multi-organ Timothy syndrome and isolated LQT8. Less than 0.5% of cases are type 8. Mutations in the CACNA1C gene–encoding for the major Ca2+ channel of the heart–may exhibit a variety of clinical manifestations. These include typical or atypical Timothy syndromes (TS) which are associated with multiple organ manifestations, and cardiac involvement in form of malignant arrhythmias, QTc prolongation, or AV block.